Sly Sendromu (MPS Tip VII)
MPS Tip VII diğer adıyla Sly Sendromu, β-glukuronidazı kodlayan GUSB genindeki mutasyonların neden olduğu nadir bir hastalıktır. Eksik β-glukuronidaz aktivitesi, dokularda sistemik GAG birikimine ve ilerleyici çoklu organ fonksiyon bozukluğuna yol açar. B-glukuronidaz enzim eksikliğine bağlı olarak progresif seyreden bir nörometabolik hastalıktır. 1,2 Lizozomlardaki GAG (Glikozaminoglikan) birikimiyle beraber çoklu organ yetmezliği, bilişsel bozukluk oluştuğu gibi yaşam beklentisi de düşüktür.1,4
Hastaların büyük çoğunluğu yaşamın ilk evrelerinde hidrops fetalis (anne karnındaki ya da yeni doğmuş bebeğin dokularında, organlarında ve vücut boşluklarında yüksek miktarda sıvı birikmesi sonucu ortaya çıkan bir sağlık sorunudur) sebebiyle kaybedilir1, semptomları daha hafif olanlar ise tanısız kalır.4
Düşük enzim aktivitesi ile dokularda progresif olarak GAG (Glikozaminoglikan) birikmeye devam eder, morbidite ve mortaliteyi ciddi anlamda etkiler. 1,4 Hastaların yarısı 1 yaşından önce kaybedilir, 15 yaşından fazla yaşayan hasta oranı ise hastaların sadece % 15’ idir.1 Hayatta kalanlar ise sonrasında akciğer yetmezliği, obstrüktif akciğer hastalığı, aspirasyon ve kalp krizinden kaybedilmektedir. 1
Genel olarak, hafif veya orta dereceli semptomları olan hastalarda kaba yüz görünümü, korneada bulanıklaşma ve sık üst solunum yolu enfeksiyonları görülür ancak hafif iskelet anormalliklerine sahiptir.1 Daha şiddetli fenotipleri olan hastalarda, boy kısalığı ve daha fazla iskelet displazisi, makrosefali, tekrarlayan kulak enfeksiyonları, gingival hipertrofi, hepatosplenomegali, fıtık ve kognitif bozukluk görülür.1 Belirtileri hafif veya orta derecede olan hastalar, daha şiddetli klinik tabloları olan hastalara göre daha yavaş kötüleşme eğilimi göstermektedir.1
Omurga ve göğüs duvarının iskelet deformiteleri yaygındır ve yaşla birlikte yavaş yavaş ilerlemektedir.1
Hastalığın infantil veya adolesan formu olan hastaların yaklaşık yarısı (%46) yardım almadan kendi başlarına yürüyebilmiştir. Bununla birlikte, çoğu hasta kalça displazisi nedeniyle bu yeteneği zamanla kaybetmiştir.1
İlerleyen iskelet sistemi deformiteleri solunum sistemi fonksiyonlarının bozulmasına yol açarak 10’ lu yaşlarda hastaların tekerlekli sandalyeye bağımlı olmasına sebep olmaktadır. 1
MPS VII’ li hastaların %50′ sinden fazlasında kalp kapak hastalığı vardır.1
İlk kez 1973’ te3tarif edilen hastalığın 300.000’ de 1 ile 2.000.0000’ da 1 sıklıkta olduğu belirtilmektedir.1
| Belirtiler1 |
| · Zeka geriliği (Ciddi belirtiler %86) |
| · Kısıtlı kelime bilgisi (Ciddi belirtiler %94) |
| · Yanlara doğru kafada genişleme (Anormal kafatası) |
| · Kaba saçlar (Yüz ve sırtta aşırı tüylenme) |
| · Kaba yüz hatları, kısa, geniş ve kalkık burun, düz yüz |
| · Büyük dil |
| · Büyük göz kapakları |
| · Korneal opasiteler, bulanık ve zayıf görme |
| · Küçük, kırılgan ve ayrık dişler, dental hipertrofi |
| · Otitis media ve işitme kaybı |
| · Kısa boyun |
| · Pectus karinatum (Güvercin, çıkık göğüs kafesi) |
| · Pectus excavatum (Göğüs kafesinde çukur oluşması) |
| · Kifoz (Yamuk omurga) ve gibbus (Kambur görünüm omurganın sırtın üst tarafında aşırı çıkması) |
| · Eklemlerde hareket kısıtlılığı (omuz ve kollarda hareket kısıtlılığı) |
| · Pençe şeklinde kaba eller (Parmaklar sertleşir ve eğrilir) |
| · Çarpık bacaklar (Genu valgum) |
| · Kısa boy (18-24 aydan sonra) |
| · Solunum ve akciğer enfeksiyonları ve ciddi uyku apneleri gözlemlenir |
| · Kardiyomiyopati ve kapak hastalıkları (%50 oranda vardır) |
| · Non immun Hidrops fetalis ( LDH’ da en sık görülen semptomdur. MPS 7 hastalarının ise %40′ ında görülür) |
| · Hepatosplenomegali (karaciğer ve dalak büyümesi) |
| · Umbilical herni (göbek fıtığı) |
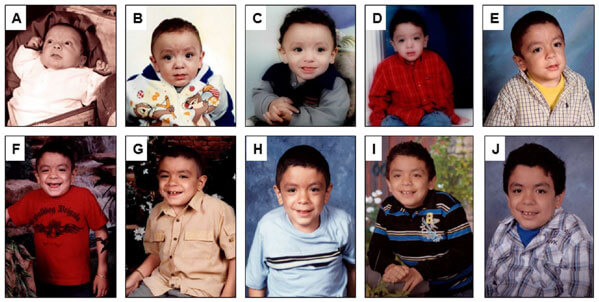
MPS VII’ de semptom başlangıcı ile tanı arasındaki değişken süre gözlenir. MPS VII teşhisi, klinik öykü, laboratuvar çalışması ve enzim/genetik testlerin bir kombinasyonunu içerir.
Herhangi bir MPS tipine ilişkin yüksek klinik şüphe, doğru teşhisin sağlanmasına ve bir genetik veya metabolik uzmanına erken sevk edilmesine yardımcı olmak için kapsamlı bir inceleme gerektirir.
MPS VII için pre ve neonatal testler erken tanıya ve müdahale fırsatına yol açabilir.
MPS VII tanısı, idrarda veya amniyotik sıvıda aşırı GAG’ ın saptanmasına, enzim aktivite ölçümü analizlerine ve genetik testlere(GUSB geni analizi) dayanır.8
Tedavi
İnsan β-glukuronidazın rekombinant bir formu olan intravenöz vestronidaz alfa (Mepsevii™) ile enzim replasman tedavisi (ERT), pediatrik ve yetişkin hastalarda mukopolisakkaridoz VII tedavisi için onaylanan ilk hastalığa özgü tedavidir. 9
Önemli Not: MPS ve benzeri lizozomal hastalıklarıyla ilgili olarak sitemizde yer alan derlemeler, gönüllü hekimlerimiz tarafından, uluslararası kaynaklar referans alınarak yapılmıştır. Amacımız, hasta ve hasta yakınlarını, lizozomal depo hastalıkları hakkında bilgilendirmek ve hastalık farkındalığını arttırmaktır. Sitemizde yer alan bilgiler, her ne kadar tıbbi bilgi niteliği taşısa da sadece bilgilendirme amaçlıdır; kesinlikle tanı, tedavi veya klinik kararlar için kullanılmamalıdır!
Referanslar:
- Montaño et al. J Med Genet. 2016; 53:403;
- Zielonka et al. Genet Med. 2017. doi: 10.1038/gim.2017.10;
- Sly et al. J Pediatr. 1973; 82:249
- Muenzer. Rheumatology (Oxford). 2011; 50(suppl 5):v4;
- P. Harmatz et al. Molecular Genetics and Metabolism 123 (2018) 488–494
- Fox et al. Mol Genet Metab. 2015 February ; 114(2): 203–208
- Lehman et al. Rheumatology (Oxford). 2011;50(suppl 5):v41;
- Muenzer. Rheumatology (Oxford). 2011;50(suppl 5):v4.
- Vestronidase Alfa: A Review in Mucopolysaccharidosis VII
Emma H. McCafferty, Lesley J. Scott BioDrugs. 2019; 33(2): 233–240.
