Maroteaux-Lamy Hastalığı (MPS Tip VI)
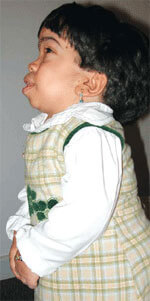
Tanımı:
MPS VI ya da Maroteaux-Lamy, N-asetilgalaktozamin 4-sülfataz(Arilsülfataz B) enziminin çok az bulunması ya da hiç bulunmaması durumunda görülen ender bir hastalıktır. N-asetilgalaktozamin 4-sülfataz normalde hücrede lizozom adı verilen kesecikler içinde bulunan ve glikozaminoglikanlar (GAG) olarak bilinen birleşik şeker zincirlerinin parçalanmasında görev alan bir enzimdir. MPS VI hastalığı bulunan kişilerde, GAG’nin birikmesi sonucunda hastalığın belirtileri ortaya çıkar. GAG’ ler deri, kemik ve organların yapısında çok önemli yapı taşlarını oluşturmaktadır.
MPS VI çok ender görülür ve sanayileşmiş ülkelerde yaklaşık 1.100 hasta bulunmaktadır.
Belirtileri:
Vücutta biriken GAG miktarı arttıkça hastalığın belirtileri giderek belirginleşir. Diğer MPS’ lerde olduğu gibi MPS VI hastalığı için tipik tek bir bulgu ya da belirti bulunmamaktadır. Belirtiler GAG’ lerin biriktiği hücrelere göre çok geniş bir başlangıç, şiddet ve tip yelpazesinde ve çocukluk, adolesan ya da erişkin dönemde ortaya çıkabilir.
İskelet belirtileri olarak, boy kısalığı, kemik anormallikleri, eklem sertliği ve yüz hatlarında kabalaşma sayılabilir. Büyüme tipik olarak 10 yaşına kadar durur.
 Organlara özgü belirtiler karaciğer ve/veya dalakta büyüme, şişkin karın, fıtıklar ve kalpte üfürümler olarak sıralanır. Diğer belirtiler arasında soluk alma güçlüğü, sesli soluma ve kulak ve sinüs enfeksiyonlarında artış bulunabilir. MPS VI hastalığının sık görülen belirtilerinden birisi de katarakttır. Hastalarda körlük ve felç oluşabilir.
Organlara özgü belirtiler karaciğer ve/veya dalakta büyüme, şişkin karın, fıtıklar ve kalpte üfürümler olarak sıralanır. Diğer belirtiler arasında soluk alma güçlüğü, sesli soluma ve kulak ve sinüs enfeksiyonlarında artış bulunabilir. MPS VI hastalığının sık görülen belirtilerinden birisi de katarakttır. Hastalarda körlük ve felç oluşabilir.
Çoğu MPS hastalığının aksine, MPS VI zekayı olumsuz etkilemez.
Kalıtım Özellikleri:
MPS VI otozomal çekinik bir hastalıktır.
Tanıya Yönelik Testler:
MPS VI tanısı genelde geç ya da yanlış konmaktadır. MPS VI tanısında kullanılan iki test bulunur. İdrar testiyle enzimdeki idrardaki GAG miktarı araştırılır. MPS VI hastalarının idrarında anormal ölçüde fazla miktarda GAG bulunur. Yapılan kan testiyle ya da deri biyopsisinden alınan örnekten çoğaltılan bağ dokusu hücrelerinde N-asetilgalaktozamin 4-sülfataz enziminin miktarı ortaya konarak hastalık tanısı kesinleştirilir. MPS VI hastalarının hücrelerinde bu enzimin miktarı çok düşüktür.
Hastalığın doğumdan önce tanısında anne karnındaki çocuğun N-asetilgalaktozamin 4-sülfataz enzimi aktivitesini araştırmak üzere tarama testi yapılabilir.
Tedavisi:
Galsülfaz ile enzim replasman tedavisinden (ERT) önce, klinik yönetim destekleyici bakım ve hematopoetik kök hücre nakli ile sınırlıydı. Galsülfaz artık yaygın olarak mevcuttur ve spesifik bir tedavidir.
Ayrıca kalıcı doku hasarını engellemek, belirtilerin ilerlemesini geciktirmek ve hasta bireylerin yaşam kalitesini yükseltmek için uygulanabilen çok sayıda uygulama bulunmaktadır.
Eklem hareketliliğini düzeltmek için genellikle fizik tedavi uygulanır. İskelet anormalliklerini düzeltmek, solunum güçlüğünü, süreğen kulak enfeksiyonlarını azaltmak üzere bademcikleri almak ve görme sorunlarını gidermek için ameliyatlar gerçekleştirilebilir.
Önemli Not: MPS ve benzeri lizozomal hastalıklarıyla ilgili olarak sitemizde yer alan derlemeler, gönüllü hekimlerimiz tarafından, uluslararası kaynaklar referans alınarak yapılmıştır. Amacımız, hasta ve hasta yakınlarını, lizozomal depo hastalıkları hakkında bilgilendirmek ve hastalık farkındalığını arttırmaktır. Sitemizde yer alan bilgiler, her ne kadar tıbbi bilgi niteliği taşısa da sadece bilgilendirme amaçlıdır; kesinlikle tanı, tedavi veya klinik kararlar için kullanılmamalıdır!
https://www.orpha.net/consor/cgi-bin/Disease_Search_Simple.php?lng=EN&diseaseGroup=Maroteaux-Lamy
https://www.mpsreference.eu/tr-tr/management-and-treatment-of-mps/mps-vi-maroteaux-lamy-syndrome/
